
















Project 001: PRYOXD1
- Date 8/13/2021
- Category Pediatric, Rare, Donate, Life Altering, Life Threatening, PryoXD1
- Tags Donate, Advocate, Communicate
Project 001: PYROXD1 – Take Part In Nat G’s Hope
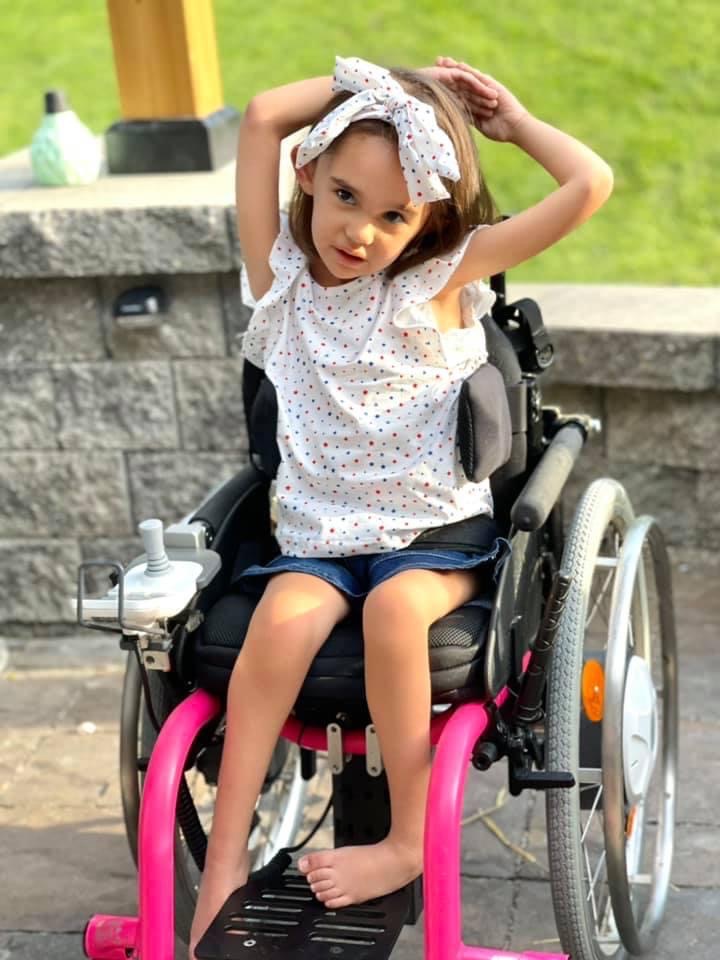
Meet our Warrior – Nat G!
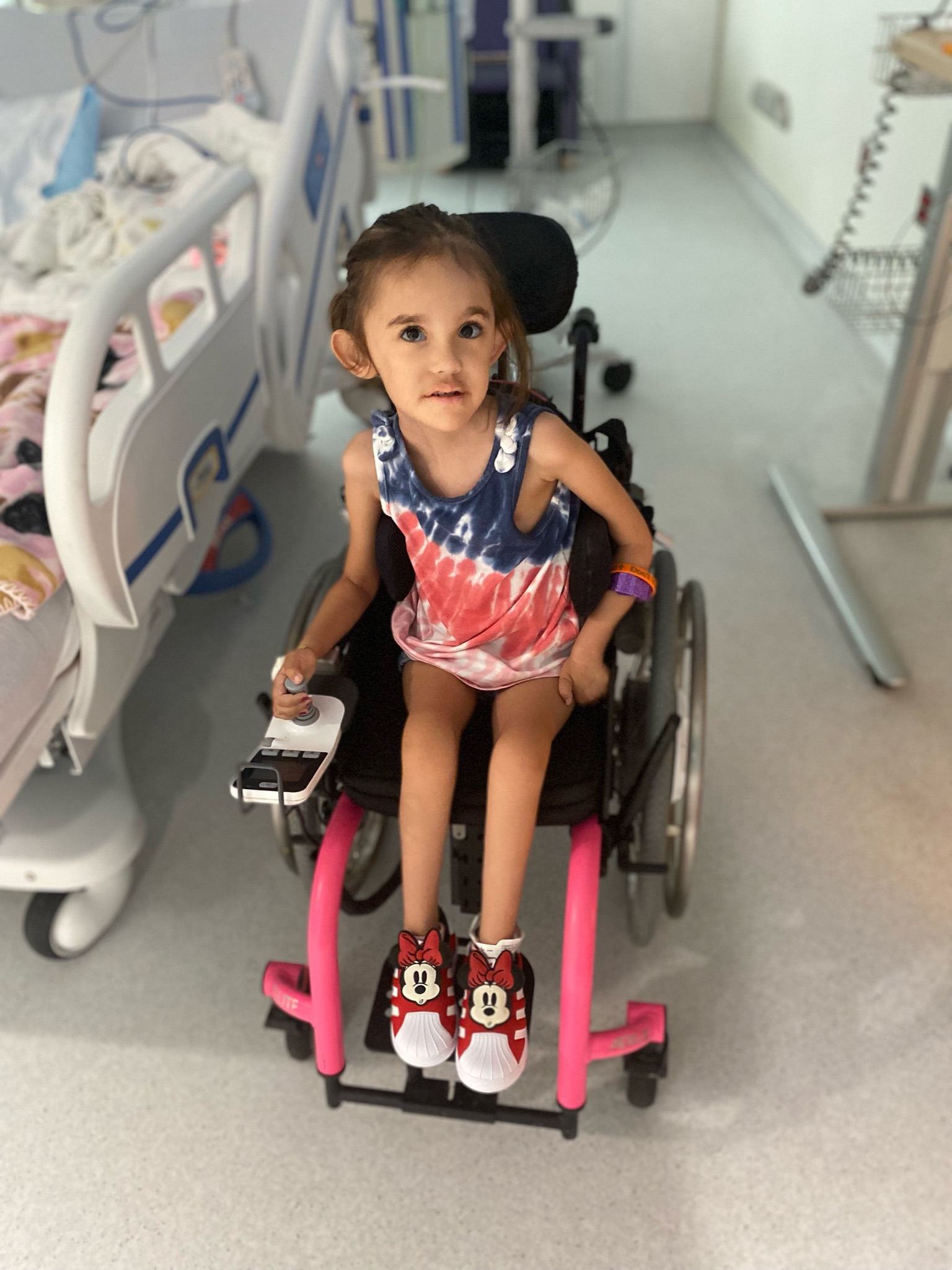
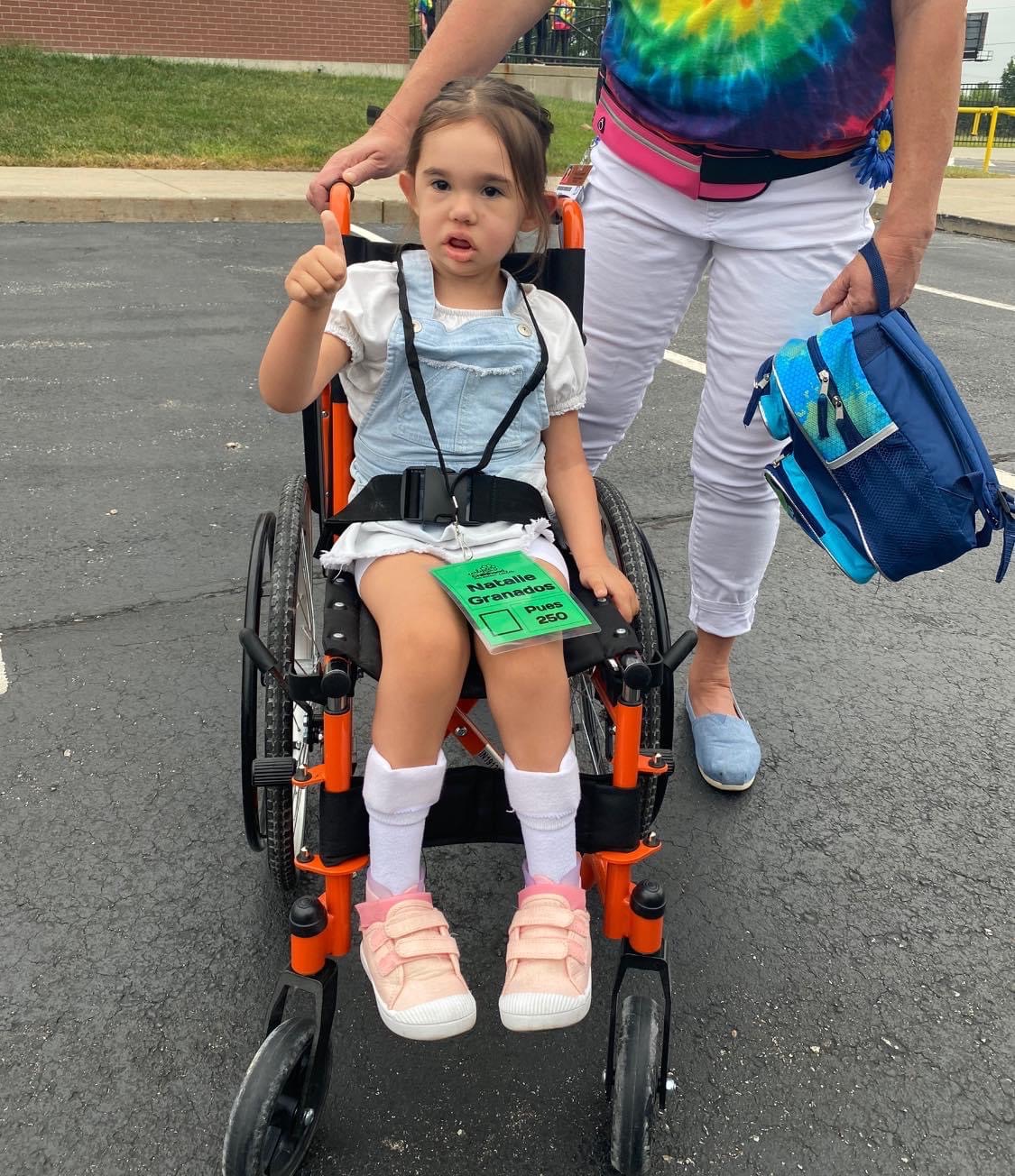
-4-year-old goofball who loves superheroes, school, and her best friend Lincoln.
-Nat G was born in 2017 and was quickly deemed a normal but “floppy” baby who would grow out of that “very soon”.
-At about 12 months old, we started physical therapy because Nat G didn’t seem to hit many “gross motor” milestones after sitting up.
-Fast forward to almost 2 years old and she was testing at 5-6 years old intellectually and with her language (basically we couldn’t shut her up! ha!).
-She was still stuck at 9 months physically.
-In Early 2020 just shy of 3 years old, and after a few layers of genetic layers.
Nat G’s Diagnosis
Myopathies are a group of genetically heterogeneous conditions characterized by muscle weakness, with overlap in the clinical presentation and histopathological features of different genetic subtypes.1 Within this group, congenital myopathies are most commonly characterized by hypotonia and weakness, often from birth, commonly with the presence of facial weakness, with or without ptosis and ophthalmoplegia.
PYROXD1 gene homozygous mutation is a very rare disease that appears to exhibit a LGMD-like phenotype with childhood or adulthood disease onset. It has few additional clinical features including scoliosis, high arched palate, nasal speech, and joint hypermobility. Awareness of these clinical features may be helpful to facilitate the diagnosis and lead to the appropriate genetic testing.
PYROXD1 is an oxidoreductase – which sounds complicated, but just means it can oxidise and reduce ‘things’. When we discovered PYROXD1, there was not a single science paper published on it, ever. We have been working from ground zero to try and find out what it does. We know that no other enzyme can reduce/oxidise ‘the thing(s)’ that need PYROXD1 – and whatever the things are, they are absolutely critical, because cells and mice do not survive without any PYROXD1.
Nat G’s Hope
The team of doctors and researchers were the first group to discover PYROXD1 as a novel disease gene causing myopathy in five families around the world. O’Grady et al, American Journal of Human Genetics 2016. They are aware of almost all cases around the world. They have been studying PYROXD1 for 7 years now – trying to find out what it is that PYROXD1 does that humans need so desperately and working our way toward a therapy.
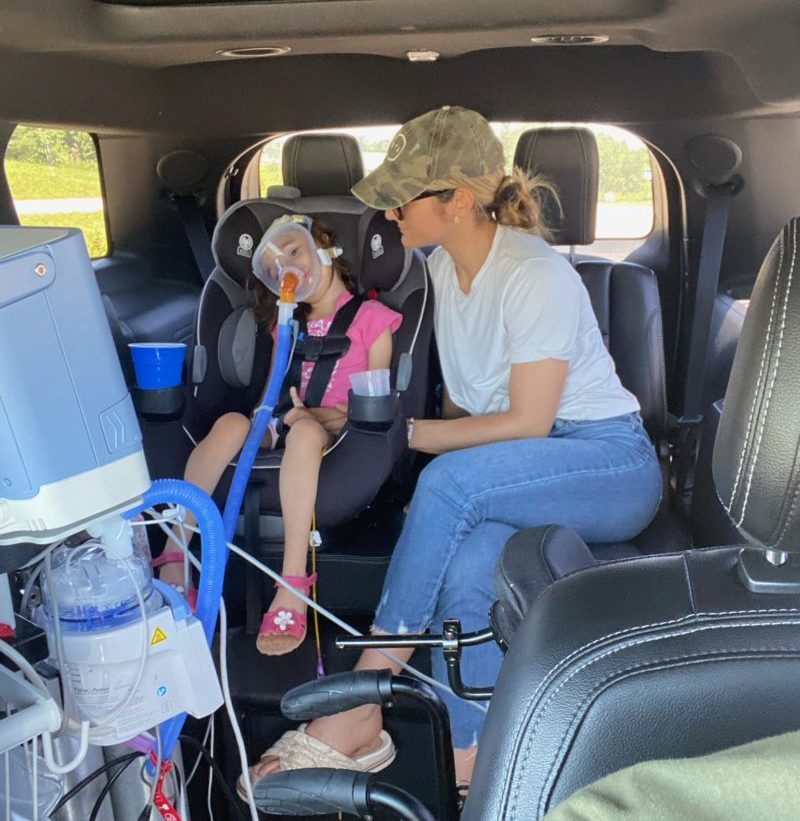
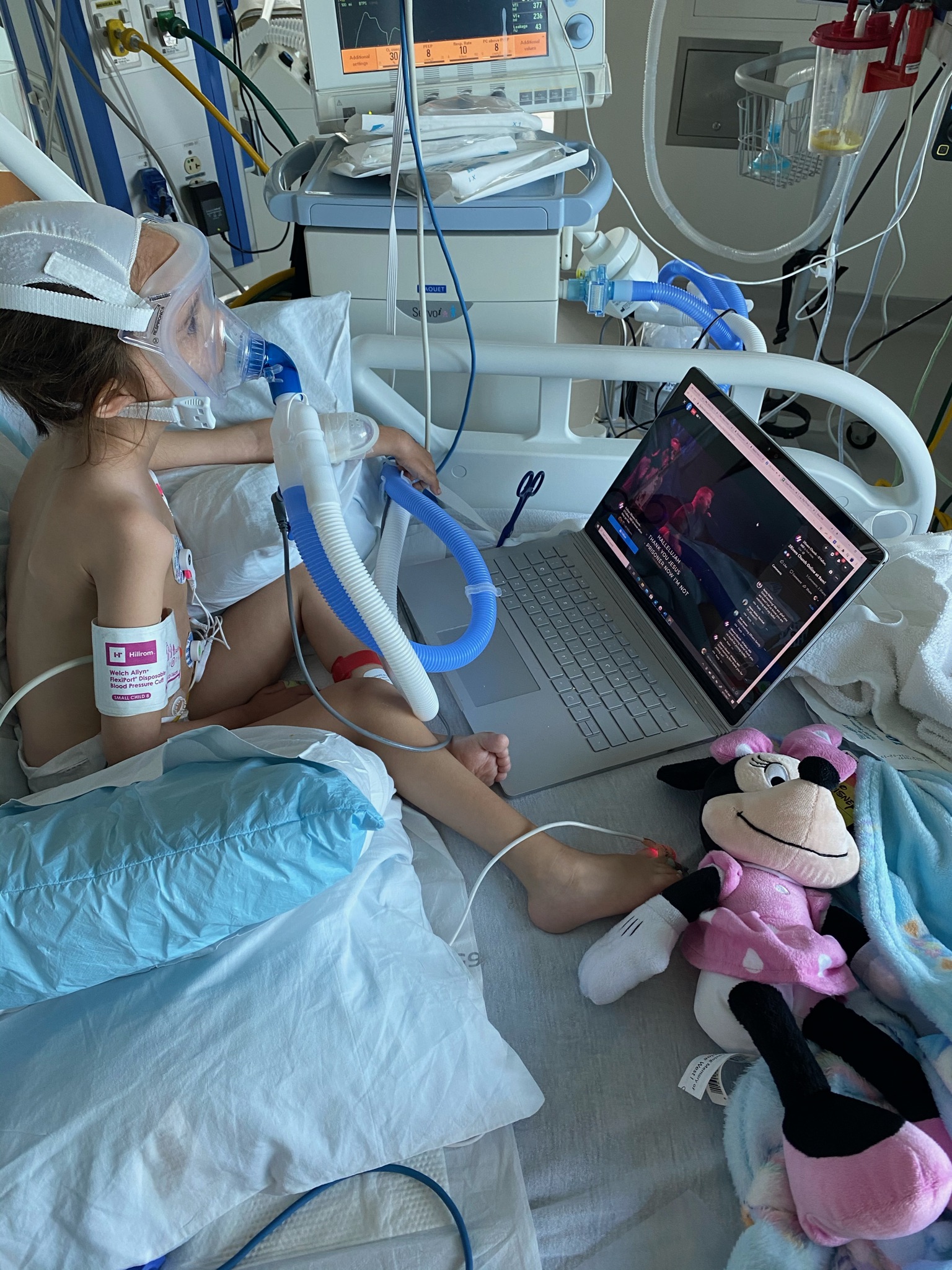
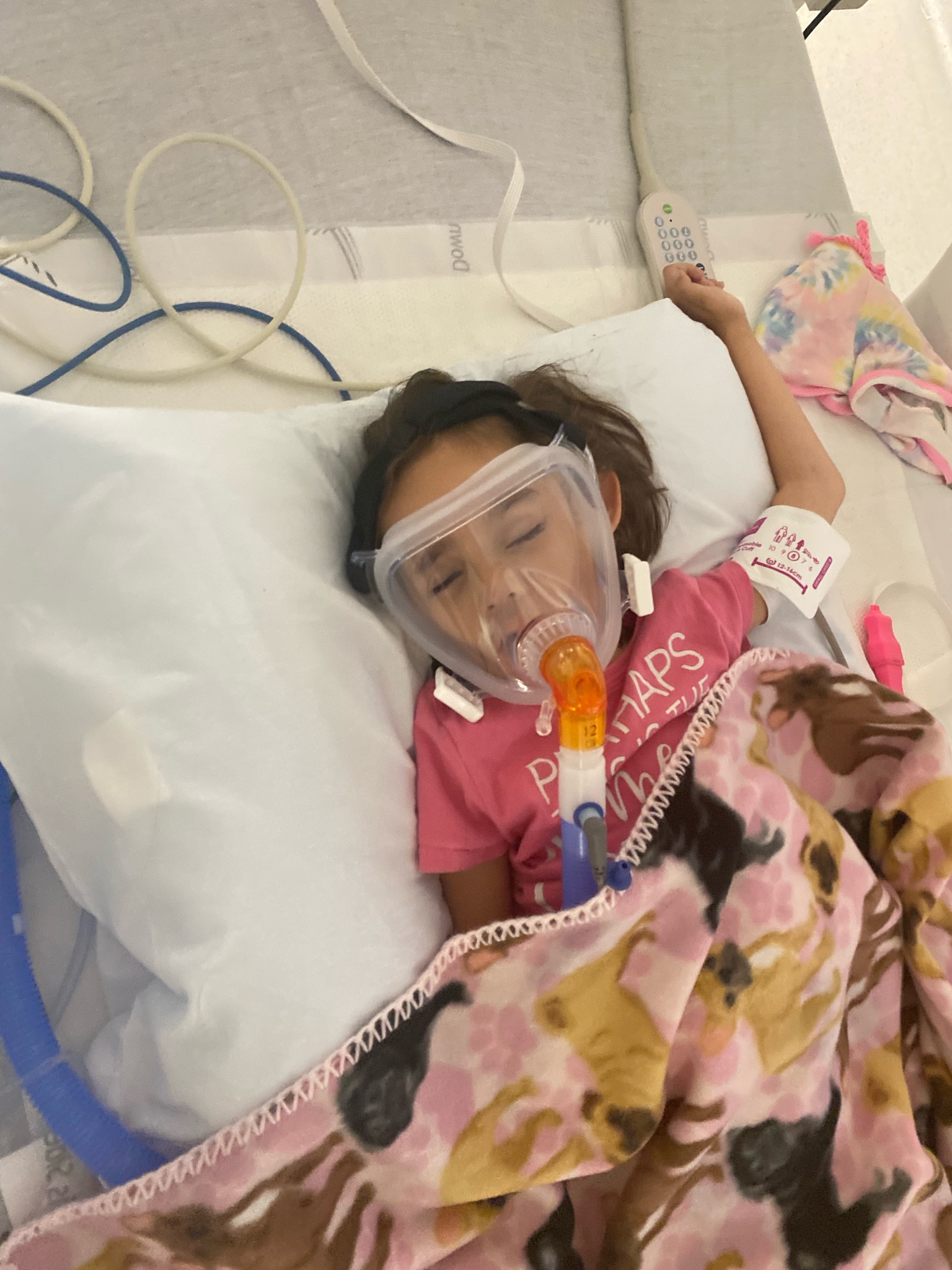
PYROXD1 (Pyridine Nucleotide-Disulphide Oxidoreductase Domain 1) is a Protein Coding gene. Diseases associated with it present as extremely rare and progressive forms of muscular dystrophy. Less than 20 people worldwide have this specific mutation, one of those people is our daughter Natalie. Her case is considered extremely severe, as her symptoms set in at a very young age. Natalie cannot walk, crawl, or do any of the things any other 3-year-old can do. She can barely use her muscles at all. A simple cold means endless trips to the emergency room. Natalie needs constant round-the-clock care, receives hyperbaric oxygen therapy twice a week ,and uses a G tube to eat. She fights every day.
Her mutation has no cure. And, due to the small number of cases, traditional research funding from pharmaceutical companies and others simply doesn’t exist. It doesn’t make financial sense.
Funding for this project, estimated at $280,000, will allow researchers to discover insights into the origin of this mutation, how it affects the body and possible treatment options. We know this won’t save Natalie, but we hope it will lead the way for finding a cure for PYROXD-1 and help others with rare pediatric diseases.
What will be done in this Project:
Researchers are trying to identify precisely what PYROXD1 does that is essential for cellular life. They know that PYROXD1 modulates the reduction-oxidation (redox) state of a vital ligand in a way that no other redox enzyme can compensate for. Many necessary enzymes likely rely upon this ligand for their activity – which explains why cells grown in the lab die about seven days after their supply of PYROXD1 is ‘genetically turned off’.
The first step in understanding what PYROXD1 does, is to create a high-resolution 3D structure of the enzyme, with and without its cofactors (FAD, NADPH), which scientists know are important for its function. Using X-ray crystallography and cryo-electron microscopy (cryo-EM) scientists can visualise the PYROXD1 molecule, map where the cofactors bind, and map the active site of the PYROXD1 enzyme where any unknown ligand could bind. By creating an accurate 3D model of PYROXD1, they can use supercomputing techniques to model what sort of chemicals can ‘fit’ into its active site.
They hope that by understanding the structure of the PYROXD1 enzyme better, the researchers will be able to design effective treatments. One of the most promising parts of this project is that the PYROXD1 enzyme can be readily made in very large quantities: the purified enzyme is soluble in water and appears to maintain potent redox activity in a test-tube. The high solubility of human PYROXD1 raises the possibility of testing enzyme replacement therapy using cell and animal models of disease, working side-by-side with doctors worldwide who are caring for families affected by PYROXD1 myopathy.
The overarching goals of the PYROXD1 researchers are as follows:
- Find out what PYROXD1 looks like – where the redox cofactors bind and map the size and shape of the active site that binds the ‘unknown ligand(s)’.
- To see precisely how patient variants affect the 3D structure of PYROXD1.
- To use computer modeling and drug screening to find chemicals that ‘fit’ into PYROXD1 ligand and co-factor binding sites that may modulate its activity.
- To test if PYROXD1 enzyme replacement therapy holds any promise using their cell and animal models of disease.
- To define the range of clinical symptoms associated with PYROXD1 myopathy and the clinical course of disease.
- To identify clinical biomarkers through non-invasive testing of blood or urine that can be to help diagnose and monitor progression of PYROXD1 myopathy (and prospectively any improvement with interventions).
The researchers goal is to use donated funding from Take Part to collate a body of strong preliminary data that will help them attract a large, federal NIH grant and publish their results in medical journals. Research discoveries along the way are likely to also have broader impact on other rare conditions, due to the fact that a ‘redox problem’ is a core pathway involved in lots of different neurodegenerative disorders.
The ideal outcomes:
- Researchers will use the structural data to identify existing drugs that may compensate for a patient’s genetic error in their natural PYROXD1. A similar approach was successful with cystic fibrosis, but they first needed a 3D structure of the cystic fibrosis chloride channel. A vital step is identifying the natural ligand(s): What binds in the PYROXD1 enzyme binding pocket? Finding this answer will be an absolute game-changer. It will also very likely inform what other therapeutic chemicals might modify PYROXD1’s activity.
- Researchers will obtain important preliminary evidence as to whether PYROXD1 enzyme replacement therapy shows any benefits in cell and animal models of disease. While enzyme replacement therapy may not work, they have to try! Therapeutic promise will help our warrior Nat G and other affected families around the world.
What are the financial needs and objectives?
If the researchers do not secure the needed funding, they could lose lots of important progress made over the last 5 years making cell and animal disease models. Most American research labs operate on a small-business model and like any small business, they need to find a way to get working capital. Unlike a small business, they are not selling anything, they are searching for something. The research teams who discovered PYROXD1 have already attracted research grants to pay labor costs supporting their staff and students and 5 years of laboratory reagents required to develop informative cell and animal models of PYROXD1 myopathy. Without ongoing funding, all of this stops.
With your help, we can provide a financial buffer so researchers can stop worrying IF further grant applications are successful, and instead can focus solely on this project. Take Part will be able to cover expenses related to these tests, as well as allow the researchers to collect sufficient data to write a full NIH grant. Through Take Part, researchers will have the confidence to continue at full speed and get themselves into the strongest position to attract federal funding in the form of 5-years of steady funds which will be used to tackle bigger scientific questions.
Financial Break Down:
Time Frame – 2 years of Research
Budget – $280,000
Below you can see the benchmarks our researchers intent to hit for each $25,000 we are able to donate.
$0-$25,000 –US Lab: Secure personnel and consumables to produce large quantities of recombinant PYROXD1 enzyme to send to the Australian lab. 3D structure research via X-ray crystallography and Cryo-EM.
$25,000-$50,000 – Australian Lab: Support laboratory costs for pre-clinical evaluation of PYROXD1 enzyme replacement therapy using Cell Models. Support early studies in Mouse Models to check the best mode of delivering PYROXD1 (oral, injected) to muscles (and also checking liver and other organs). Detailed analysis of disease progression in the mouse model. Continued research into clinical biomarkers of PYROXD1 disease in affected patients.
$50,000-$75,000 – US Lab: Make and purify different versions of PYROXD1 enzyme to act as important controls for Cell and Mouse studies. These might be an “enzyme-dead” version to act as a negative control or patient versions of PYROXD1. Chemical screens of therapeutic agents.
$75,000-$100,000 – Australian Lab: Support personnel and laboratory costs for: 1) more detailed Pre-Clinical evaluation of enzyme replacement therapy in cell models using different versions of PYROXD1 made by the US lab; 2) Preliminary dose and toxicity trials in Mouse Models; 3) Studies of mice housed with an exercise wheel with sophisticated electronic monitoring to test if voluntary daily exercise improves or worsens disease progression and if mice with PYROXD1 myopathy run the same distance or speed as their littermate controls that don’t have PYROXD1 myopathy.
$100,000-$125,000 – US Lab: Support personnel and laboratory costs to make and purify enough PYROXD1 enzyme for a pre-clinical trial in mice. Make and purify a range of different patient-based PYROXD1 genetic variants. Deploy a suite of biochemical assays to study the behavior of wild-type and patient PYROXD1 enzymes and any modulatory effects of ligands, co-factors and therapeutic chemicals on PYROXD1 enzymatic activity.
$125,000-$200,000 – Australian Lab: Support personnel and animal costs for a pre-clinical trial of enzyme replacement therapy in the PYROXD1 mouse model, with and without the running wheel (including purchase of additional running wheels). Animals will need constant monitoring over ~ 2-3 month treatment window and it is possible the dosing regime may require weekly injections.
$200,000-$225,000 – US Lab: Leverage information from structural 3D models to hone in upon chemicals most likely to ‘fit’ into the PYROXD1 active site and modulate its activity. Expand biochemical assays to conduct detailed analyses of ‘the strongest modulators’ of PYROXD1 activity for a range of different patient-based forms of PYROXD1.
$225,000-$250,000 –Australian Lab: Blinded analyses of tissue phenotypes, running data and health and weight data from the enzyme-replacement trial in the PYROXD1 mouse model. This lab are expert in neuromuscular disorders and will perform a suite of phenotypic investigations that quantify disease severity. Compile all findings from cell and animal models to apply for a much larger grant to conduct more expansive animal studies to refine dosing and treatment regime(s).
$250,000-$280,000 – US Lab: Compile all findings from 3D structure X-ray crystallography, CryoEM and biochemical assays to apply for a much larger grant to test therapeutic benefits of pharmacological factors shown to modulate PYROXD1 activity in cell and animal models. Bring laboratory findings into a therapeutic option for affected families.
Three Ways You Can Take Part:
- Donate: Take time right now to donate to this project or Take Part as an organization so we can bring more money to the researchers
- Advocate: Take time right now to share this project through social media and any other way to let others know they can help this worrier and many others who have been (and will be) diagnosed with this condition
- Connect: We are always looking for corporate partners who would be willing to come along side us and donate for these causes. Some corporate sponsors donate parts of project and other will donate the entire amount to help researcher to focus on the research. If you work for a company who you feel may be able to donate to this cause, please click here to contact us and let us know you are interested. We will walk you through the entire process so it is as easy as possible for you.
The researchers Take-Part chooses to support are responsible for the direction of where the funds are going. so the plans above have been designed by the researcher with no influence from any donors. When it comes to medical research, we need the researchers to be able to pivot as needed in their research as findings are discovered. The above is what the researchers have shared they intend to use funds raised to achieve the desired goal of this project. Take-Parts role in this is to help caring donors like you, put funds towards amazing researchers that we decide to support, and allow the researchers to do what is best for the progression of this project.